Hello and welcome to our March 2017 newsletter. In this issue, we discuss Brexit and its implications for the UK market, how managed entry agreements are now back in favour with both payers and manufacturers and how has the UK’s Cancer Drug Fund performed under NICE’s stewardship?
We will be presenting at a number of upcoming events, including the Global Market Access Forum in Frankfurt on 4th May. We will also be presenting at ISPOR in Boston on 20-24th May on “Is NICE being NICE in their re-evaluation of oncology drugs in the Cancer Drugs Fund in England”. It will be great to catch up with you there.
Finally, we are growing and are actively searching for talented people to join our team for a number of open positions. Please do contact us if you have any suggestions or recommendations.
Brexit – Into the unknown for the UK, EU and patient access to pharmaceuticals?
On the 29th March 2017, the UK government triggered Article 50, which initiated the negotiations for the UK to withdraw from the European Union. The UK and EU now have two years to reach an agreement as to how the UK can exit the EU and what the terms will be for the new relationship with the EU. For the next two years, nothing is likely to change for patient access of pharmaceutical products in the UK, it does pose interesting questions for 2019 and onwards.
What we know
The UK government has stated that it is its intention to leave the EU single market, customs union and the European Court of Justice (ECJ). This implies that the UK is heading for a so called “hard Brexit”, which will involve a new trade deal being negotiated. Whether this new trade deal will result in comparable access for the UK to the EU single market remains to be seen and will only become clear as the negotiations progress.
What are the potential implications for pharmaceuticals and patient access from 2019?
This can be broken into 3 topics: The EMA and market authorisation, the UK pricing and market access process and the UK’s role within EU pharmaceutical industry.
1. The EMA and marketing authorisation (MA)
If the UK is no longer part of the single market nor under the jurisdiction of the ECJ, it is highly probable that it will leave the EMA. The implication is that the UK would no longer be part of the joint EU marketing authorisation process. This would require a separate marketing authorisation process for the UK, presumably managed by the MHRA. This will probably require additional cost, time and resource implications for manufacturers and will result in a delay in seeking marketing authorisation to the UK market. It is possible that an agreement could be negotiated in which the UK MA process is classed as comparable to the EU MA process (e.g. similar to Norway), but this remains to be seen.
Remap Consulting’s working assumption after 2019: The UK leaves the EMA and a separate regulatory process is required to gain marketing authorisation to the UK market
2. The UK pricing and market access process
Given that pricing and reimbursement of pharmaceuticals has been clearly stated to be the responsibility of the member states by the EU, there is unlikely to be any change in the price and funding of new pharmaceuticals, unless the UK government decides to. The result is that the Department of Health will be responsible for setting the price, whilst NICE and the representative bodies in the other devolved countries will maintain their role in deciding access and funding.
Remap Consulting’s working assumption after 2019: No change to the UK’s pricing and funding processes for gaining access
3. The UK’s role within EU pharmaceuticals
From a European perspective, the UK’s withdrawal from the single market could have major consequences, specifically on: Parallel trade, International reference pricing and Launch sequence.
- Parallel trade
If the UK withdraws from the single market and the UK does not adopt a comparable marketing authorisation framework, then parallel trade to and from the UK should cease. However, if the UK adopts a comparable regulatory regimen, it is likely that parallel trade will continue.
Remap Consulting’s working assumption after 2019: Parallel trade is unlikely to continue, due to the different marketing authorisation process.
- International reference pricing (IRP)
The UK is one of the most widely referenced countries from an IRP perspective and will continue to be in the short term, even if it leaves the EU single market. It is likely to take time for EU markets to update their IRP baskets, if they choose to do so. If there is further depreciation in Sterling, then the UK may continue to be an attractive reference country due to its relatively low prices, which will pose a challenge to manufacture’s profitability.
Remap Consulting’s working assumption after 2019: The UK will remain an important IRP market, but this is likely to pose challenges to EU and global price corridors, particularly if further depreciation in the currency occurs.
- Launch sequence
Due to the likelihood of a separate regulatory regimen, it is likely the UK will slip down the global launch sequence, due to the resource constraints faced by most countries. As such it is likely to be on the second tier of markets, such as Switzerland and Canada.
Remap Consulting’s working assumption after 2019: The UK will move to a second wave of global launch markets, due to the differing marketing authorisation process.
As negotiations have just started between the EU and UK, nothing is certain about the process nor outcomes of the UK withdrawing from the EU and there is likely to be many more surprises along the way. One top 15 pharma company recently told us that they are currently excluding the UK from market research, due to the huge uncertainty over launching within the market. They prefer to see the UK as a potential upside if the new products can be launched there. We will keep you abreast of the UK situation and it will be interesting to see how our accurate working assumptions are in late 2019!
Managed-entry agreements (MEAs) – from exception to norm?
Regularly newspaper headlines talk about struggling healthcare systems, restricted healthcare budgets and increasing treatment costs. They also highlight, particularly in Europe, that innovative treatments do not reach patients as payers deny market access. Key drivers for market access rejections are high perceived product price, large estimated budget impact and/or concerns around clinical efficacy and safety as well as patient population. Managed entry agreements (MEAs) are one potential way of reducing payer concerns and uncertainties and enabling patient access.
What are MEAs?
MEAs are also known by other names such as risk-sharing agreements, value-added services, result-base payments and patient access schemes. They are contracts between manufacturers and payers that help mitigate payer uncertainty and allow faster access to new innovative healthcare technologies. Finance- and price-based MEAs have been around for many years in various forms. However, more recently outcome-based MEAs have been introduced (Figure 1).
What are MEAs?
The manufacturer can enter:
- A finance-based MEA that addresses the payer’s budget impact concerns by reducing the net price of a product
- An outcome-based MEA that addresses the payer’s concern around the clinical value such as efficacy and safety.
Both categories can operate on an individual patient level or at the population level resulting in four subcategories (Figure 1). Within these subcategories, there are different approaches a manufacturer can adopt when introducing a MEA (Figure 1).
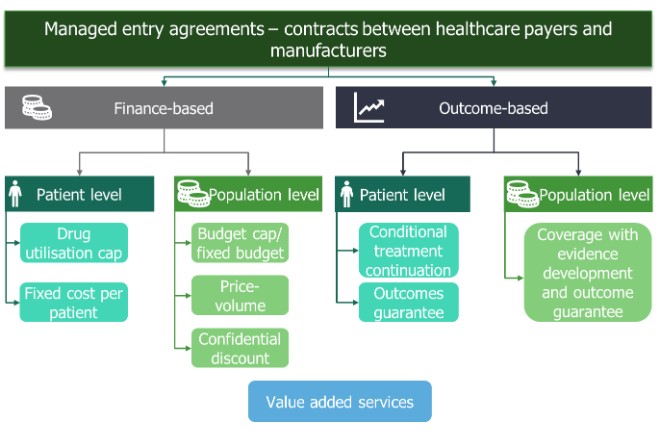
Advantages and disadvantages of MEAs
All MEAs pose advantages and disadvantages to both the manufacturer and the payer. MEAs require time and resources to develop, initiate and implement. Some MEAs (particularly outcome-based MEAS) require additional monitoring and/or data collection across the product lifecycle.
Across the globe, payers and manufacturers are more familiar with finance-based MEAs. These types of agreements are typically less complex and relatively easy to implement, particularly at the population level. Finance-based MEAs generally address the payer’s concerns around budget impact. The agreements do not affect the list price of the product and therefore do not impact prices in other countries. Manufacturers need to be careful when negotiating finance-based MEAs to mitigate the potential financial downside of the agreements.
Over the past two decades, payers and manufacturers have experimented with outcome-based MEAs with varying degree of success. Outcome-based MEAs typically link the payment the manufacturer receives for the drug to treatment success. As such, outcome-based MEAs are generally more complex to implement and execute than finance-based MEAs as they require the treatment success to be monitored. In addition, outcome-based MEAs might also require additional data collection. For payers, outcome-based MEAs reduce the risk of funding a product that provides less clinical benefit than anticipated. While for manufacturers, the financial return can increase if the product performs better than expected.
When should manufactures opt for MEAs?
Even though MEAs have become more widespread and there are many examples, MEAs should be the exception rather than the rule. Manufacturers should do their utmost to ensure that their clinical development program is robust, providing the necessary evidence for payers to evaluate the product and its value. In addition, the price should reflect the product’s perceived value.
Finance-based MEAs can be used for products with a predicted high budget impact such as:
- Products with high cost and low volume
- Products with low cost and high volume.
Outcome-based MEAs are appropriate under specific conditions:
- The new treatment is innovative and addresses a high unmet need
- The clinical benefit is associated with significant uncertainty
- The clinical benefit shown in clinical trials might not reflect the benefit in real-world clinical practice.
When developing a MEA the manufacturer needs to ensure that:
- The type of MEA is appropriate for the competitive environment and company aspirations for the product
- There is a clear rationale for the MEA
- The MEA addresses payer concerns and uncertainties
- The national/regional/local healthcare infrastructure can support implementation of a MEA.
To ensure the success of a MEA manufacturers should engage early with payers, health technology assessment (HTA) bodies and other stakeholders. They must also secure alignment within their organisations (e.g. market access, clinical, finance, health economics, legal) as a wide skillset is required to successfully implement agreements. Early preparation is a must.
In summary, adoption of MEAs is becoming more widespread, both across markets and the types of MEAs available. Manufacturers and payers see MEAs as useful tools to allow access to innovative pharmaceuticals where there is too much uncertainty over the value of the product, either from a financial or clinical perspective.
How is NICE coping with the management of the CDF?
In July 2016, NICE took over responsibility for the Cancer Drugs Fund (CDF) in England, with an annual budget of £340 million. The new process aims to be completely transparent and should enable faster patient access to oncology treatments that have the potential to be cost-effective.
Under the new CDF scheme, the intention is that NICE will evaluate all new cancer drugs and provide draft guidance prior to market authorisation. NICE will provide one of three recommendations:
- YES – Recommended. The drug is recommended for routine commissioning by NHS England.
- NO – Not recommended. NICE does not recommend the drug for use in NHS England. However, patients may still get access to the treatment through individual funding requests
- Recommended for use within the CDF. NICE consider there to be potential for the drug to be cost-effective, but there is clinical uncertainty which needs investigation, through data collection in the NHS or clinical studies. In these cases the CDF will fund the drug, for up to two years, whilst the additional data is being collected. NICE will then review the new evidence to determine if it should be recommended for routine commissioning by NHS England.
Those products with positive draft guidance will have interim CDF funding available at market authorisation. The interim funding will cover the time taken for NICE to publish its final guidance (90 days after marketing authorisation) and the time taken for baseline NHS funding to be made available (an additional 90 days).
Drugs receiving recommendation for use within the CDF are also eligible for this interim funding. This type of decision will be made for treatments that NICE consider to be genuinely promising, but do not yet have robust enough data to allow a final decision to be made. The company will sign a managed entry agreement at the time of NICE final approval with the price decided within the managed access agreement. This price is expected to reflect the uncertainty of the product. At the end of the managed access period (approximately 2 years) NICE will re-appraise the drug given the new evidence that has been generated and a final decision will be made as to whether to NHS England should permanently fund the product.
The NICE CDF assessment process is comparable to that of a single technology appraisal (STA), with the exception that the process will start prior to marketing authorisation and will last 21 weeks, as opposed to the 34 weeks for an STA. If NICE are to make a draft recommendation prior to marketing authorisation, this means that the pharmaceutical company must make their submission to NICE at least 12 weeks prior to the marketing authorisation.
At the time of reform, the existing CDF contained 25 drugs for treatment of 35 indications. In addition to evaluating all new oncology treatments, NICE are currently carrying out a rapid review assessment of the existing CDF drugs to determine their cost-effectiveness. Appraisal of all existing drugs on the CDF is expected to be complete by early 2018.
To date, NICE has evaluated nearly 50% of the drugs on the existing CDF and appears to have been quite positive, with 58% of drugs (14 of the 24) assessed by NICE being recommended for routine use in the NHS. However, for each of these drugs the pharmaceutical company has agreed to a patient access scheme or a restricted patient population in order for the drug to be considered cost-effective and accepted by NICE. It is also interesting to note that in 2015, 16 drugs for the treatment of 27 indications were removed from the CDF prior to NICE taking over responsibility for the CDF. Had these products been included in NICE’s rapid review – the NICE approval rate would be significantly lower!
Whilst the new NICE CDF approach offers a more transparent process and will enable faster access to cost-effective oncology treatments, the process there are a number of considerations pharmaceutical companies should take into account:
- Companies are likely to have to offer competitive patient access schemes to get their product to be considered cost-effective. This will have an impact on the net price.
- Companies must be pro-active and have in place a strategic and tactical real-world evidence plan if they believe the current evidence base is not sufficient for a positive NICE recommendation
- Companies will be required to submit their evidence dossiers to NICE before market authorisation. This will have implications from a company resource and data analytics perspective, it is likely that some of the data required for evaluation by NICE may not be available at time of the company submission to NICE
- Companies bear all the financial risk of the CDF as all drugs funded by the CDF will be subject to an expenditure control mechanism whereby any overspend will have to be paid back.
Interesting articles that caught our attention

R&D costs for pharmaceutical companies do not explain elevated US drug prices
Thought provoking analysis showing that the US price premium more than covers the global R&D expenditure of pharmaceutical companies https://goo.gl/PUUiXI

Roche sets disruptive price for new MS drug Ocrevus in US
Roche has adopted an unusual pricing strategy to try to gain fast market penetration at a lower price than existing competitors. It will be interesting to see how this develops https://goo.gl/ttwvtH

Drugmakers manipulate orphan drug rules to create prized monopolies
An analysis of how financial incentives for orphan drugs have increase demand and higher prices for products https://goo.gl/mWQIeA